
发布时间:2017-06-15
一直以来,医疗器械特别是高风险医疗器械的可追溯性是监管工作中着力解决的难题,无论是对不良事件监测、器械在供应链中的识别、事故器械的召回还是上市后警戒系统的建立,都具有重要的基础性影响。随着电子信息技术的发展,医疗器械唯一性标识(Unique DeviceIdentification,UDI)系统已在监管水平较高的国家和地区建立并广泛应用,目前我国也正在尝试建立适合我国国情的医疗器械UDI 系统,以加强医疗器械流通领域的可追溯性。
牙科医疗器械包含了器械、设备、器具、材料等各类型产品,其管理类别从Ⅰ到Ⅲ类均有分布,产品种类丰富情况复杂,UDI 的要求也有所不同,本文参考相关资料,对欧美相关要求做一介绍和解读。
1.欧盟最新UDI 要求
欧盟尚未对牙科医疗器械发布UDI 相关专门要求,在其即将发布实施的最新医疗器械法规(Medical DevicesRegulation,MDR)[1] 中引用国际监管者论坛(InternationalMedical Device Regulators Forum,IMDRF)[2] 相关指南文件的概念,对医疗器械UDI 作出了基本要求:
1.1 定义
“器械唯一性标识”指一系列数字或字母字符,根据国际接受的器械标识和编码标准产生,实现市场上特定器械的明确标识。
1.2 UDI 系统相关条款
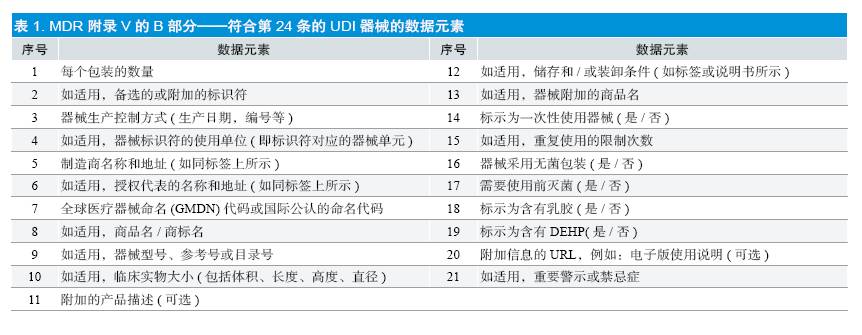
根据MDR“第24 条器械唯一性标识系统”的要求,UDI 系统应由理事会指定一个或多个实体按照本法规来运行一个UDI 分配系统,并满足以下条件:①该实体是一个法人组织;②按照本法规的要求进行UDI 的分配和使用,使其足以标识一个器械;③其分配UDI 的系统符合相关国际标准;④该实体根据预设、公开的术语和条件,使所有利益相关用户可以进出其UDI 分配系统;⑤该实体应承担以下事项:运行其UDI 分配系统一定时间,该时间在指定该实体时明确,至少为三年;根据要求,向理事会和成员国提供其UDI 分配系统相关信息,以及在器械标签上置入与实体系统一致的UDI 生产商相关信息。指定期间始终符合与指定相关的标准,以及相关条款。
MDR 同时还规定联盟中的器械,除定制式或研究用之外,应采用器械唯一性标识系统。UDI 系统应允许器械的标识和可追溯性,并由以下内容组成:①生成含有以下内容的UDI :器械标识符,对应特定制造商和器械型号,包含附录Ⅴ的B 部分信息(见表1);生成标识符,标识与器械生产单元有关数据。②在器械标签上置入UDI。③经济从业者和卫生机构以电子方式储存UDI。④建立一个UDI电子系
统。
2.美国牙科器械UDI 相关要求
FDA 建立UDI 系统的初衷在于:①使不良事件得以被更准确的报告、回顾和分析,从而使问题器械能被更快地识别和纠正。②使医疗专业人员和其他人可以更快、更准确地识别一种器械并获得该器械特性相关信息,从而减少医疗失误。③提供一种标准和清晰的方式,对电子病历、临床信息系统、索赔数据中所用器械进行建档,以促进对市场中器械的分析。一个更强的上市后警戒系统还能支持新器械的上市前审核或声明,以及已上市器械的新用途。④提供一个标准化的标识符,使制造商、分销商和医疗机构得以更有效地管理医疗器械召回。⑤为一个全球的安全分销链提供基础,帮助进行器械伪造、转移的定位,为医疗紧急事故做准备。⑥引导建立一个世界公认的医疗器械标识系统的开发。
美国是全球最早建立和应用UDI 系统的国家之一,随着法规要求的正式实施,为了保证牙科器械领域能顺利应用该系统,2015 年7 月,美国牙科协会批准发布了“美国牙科协会技术报告No.1081——FDA 关于牙科器械及作为医疗器械管理生物产品的器械唯一性标识(UDI)计划”,该报告对UDI 的基本要求做了简要介绍,并对整体要求下,牙科器械领域的具体实施要求和计划进行了解析[3]。
2.1 美国UDI 系统基本情况介绍
UDI 规则是美国FDA 起草的一项旨在识别分销链中器械的规则。该规则要求医疗器械的标签上纳入一个器械唯一性标识符(UDI)。同时贴标者必须向FDA 的全球器械唯一性标识数据库(Global Unique Device IdentificationDatabase,GUDID)提交器械产品信息。本规则建立的系统要求每个医疗器械的标签和器械包装含有一个UDI,并要求每个UDI 同时以一个纯文本和一个采用自动标识和数据获取(Automatic Identification and Data Capture,AIDC)技术的形式提供。如果该器械预期使用一次以上,且拟在每次使用前进行再加工处理,UDI 将被要求直接标在器械本身。
随着信息电子技术的发展,UDI 的记录方式呈现多样化,例如:射频识别(RFID):利用电磁场进行数据无线传输,实现物体上标签的自动识别和追踪;矩阵(2D)条形码:一个类似线型条形码的二维信息表达方式,但可以表达更多数据;线型条形码:由不同宽度的线条和空间组成的“一维”条形码,这些代码可由GS1,ICCBBA 或HIBBCC 制定。
一个产品的UDI 是一个唯一性的数字或字母代码,由两部分组成——器械标识符(Device Identifier,DI)和生产标识符(Production Identifier,PI),前者是一个UDI 强制的、确定的部分,标示出贴标者,器械特定版本和型号,后者为对应特定条件的、可变的部分,在器械标签上标示以下一项或多项内容:①器械生产所属的批号;②某一特定器械的序列号;③某一特定器械的有效日期;④某一特定器械的生产日期;⑤作为器械的人源细胞、组织或基于细胞或组织的产品(HCT/P)的特殊标识代码。
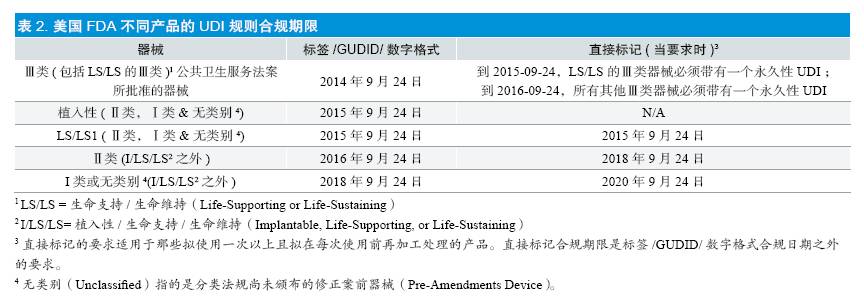
作为UDI 系统的一部分,FDA 建立了全球器械唯一性标识数据库(GUDID),其中包括了每个带有UDI 器械的基本标识元素基本信息。GUDID 接受GS1,HIBCC 和ICCBBA 的输入,且该信息的大部分面向公众公开,以便器械的使用者查到器械相关信息。UDI 没有标出,数据库也不会包含,关于谁用了器械的任何信息,以及个人隐私信息。针对不同管理类别和风险类型的产品,FDA 给出了不同的UDI 合规期限(见表2)。
2.2 牙科器械实施UDI 的时间轴
①作为最高风险的器械,FDA 要求Ⅲ类医疗器械在2014 年9 月24 日前在标签上纳入UDI,除非贴标者获批延期一年。牙科器械该类别产品包括产品代码为NQA 和NPZ 的骨替代材料等。
②牙科种植体(不论其监管类别),产品代码为NPL,NPK 和NPM 的骨替代材料,其他生命支持或生命维持器械,其UDI 合规期限为2015 年9 月24 日。植入性器械不要求直接标记。
③重复使用器械——如果器械预期使用一次以上,且预期在每次使用前进行再加工处理,则必须有UDI 永久性地直接标在器械本身。该永久性UDI 可与器械标签上的UDI 一样,也可是一个不同的UDI,以区分器械本身和其包装。永久性UDI 必须是(其中一条或两者兼具):第一,易读取文本;第二,AIDC 形式或其他向器械提供UDI 的技术。该永久性UDI 要求不适用,当该类器械符合以下任何情况:第一,任何直接标记将干扰器械的安全性和有效性;第二,器械上直接标记在技术上不可行;第三,器械声明或审批为一次性使用器械;第四,器械已有一个永久性UDI 直接标记在器械上。
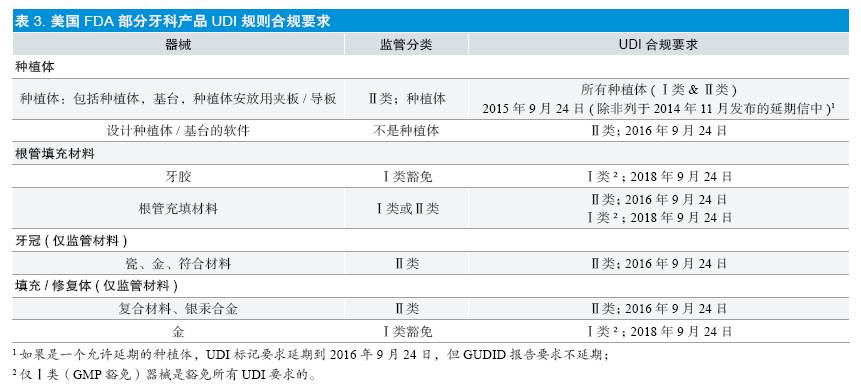
④Ⅱ类器械的UDI 合规期限为2016 年9 月24 日,Ⅰ类器械的UDI 合规期限为2018 年9 月24 日。为了帮助实施者理解,报告以列表形式给出了部分产品的UDI 实施时间要求(见表3)。
此外,FDA 向贴标者的现有存货提供了一个为期三年的延长期。如果贴标者有在器械UDI 合规期限前已完成包装和贴标的器械,他们有3 年时间使用这些存货,随后才要求所有包装和贴标的器械必须实现UDI 合规。
3.小结
每年各国监管者都收到成千上万的医疗器械报告,怀疑死亡、重伤和故障与器械有关。理想的结果是有一个终端对终端的、透明的追踪系统。随着欧盟医疗器械新法规MDR 的正式发布实施,以及美国FDA 在各个领域不断推进UDI 系统的应用,医疗器械行业采用基于现代电子信息技术的医疗器械唯一性标识系统已是大势所趋,并势必产生全球性的影响。我国在该方面的更新速度也将影响我国医疗器械行业能否及时与国际市场接轨。
牙科医疗器械种类繁多,涉及领域广泛,各品种风险程度差异较大,UDI 规则的实施更加需要具有针对性的、科学合理的具体方案。另一方面,随着社会水平的发展,人们对于口腔疾病的重视程度不断提高,就诊需求越来越大,应运而生的是开遍大街小巷的牙科诊所,这些诊所水平、层次参差不齐,这一现状给牙科器械的可追溯性带来了更加迫切需求和挑战。如何建立和应用UDI 系统来保障不同牙科器械的可追溯性,如何适应这一变化给行业带来的影响,是监管者与行业各方面都应切实思考的问题。
来源:《中国医疗器械信息》杂志

 首页
首页 
 关于医博会
关于医博会 展会概况
展会概况
 展商中心
展商中心
 观众中心
观众中心
 媒体中心
媒体中心
 网上展厅
网上展厅
 商旅服务
商旅服务
 同期活动
同期活动
 联系我们
联系我们
 丞华展览
丞华展览





